









6/13/2017
PITTSBURGH, July 13, 2017 – The scientific textbook depiction of the flu virus is about to get a facelift, due to a University of Pittsburgh School of Medicine team’s discovery that a model of the influenza genome architecture untouched since the 1970s isn’t so perfect after all.
The discovery, reported online and in a coming print issue of the journal Nucleic Acids Research, reveals loopholes in the way the virus packages its genetic material. When one strain of flu co-mingles with another strain inside a cell, these loopholes allow the viruses to swap genetic material and give rise to new strains of flu. Knowing these loopholes and how they interact with each other could give scientists the opportunity to better predict pandemics and find new ways to disrupt the flu virus.
“Although influenza has plagued mankind for hundreds of years and poses a substantial public health threat every winter, we know surprisingly little about flu pandemics,” said senior author Seema S. Lakdawala, Ph.D., assistant professor in Pitt’s Department of Microbiology & Molecular Genetics. “Our discovery may give insight into how the flu virus continually evolves, opening the door to better vaccines and antivirals.”
Influenza is a type of virus that uses single-stranded ribonucleic acid (RNA) to replicate, instead of double-stranded DNA. Influenza viruses are made up of eight RNA segments bound by a protective nucleoprotein. All eight RNA segments must come together inside a virus particle to be fully infectious.
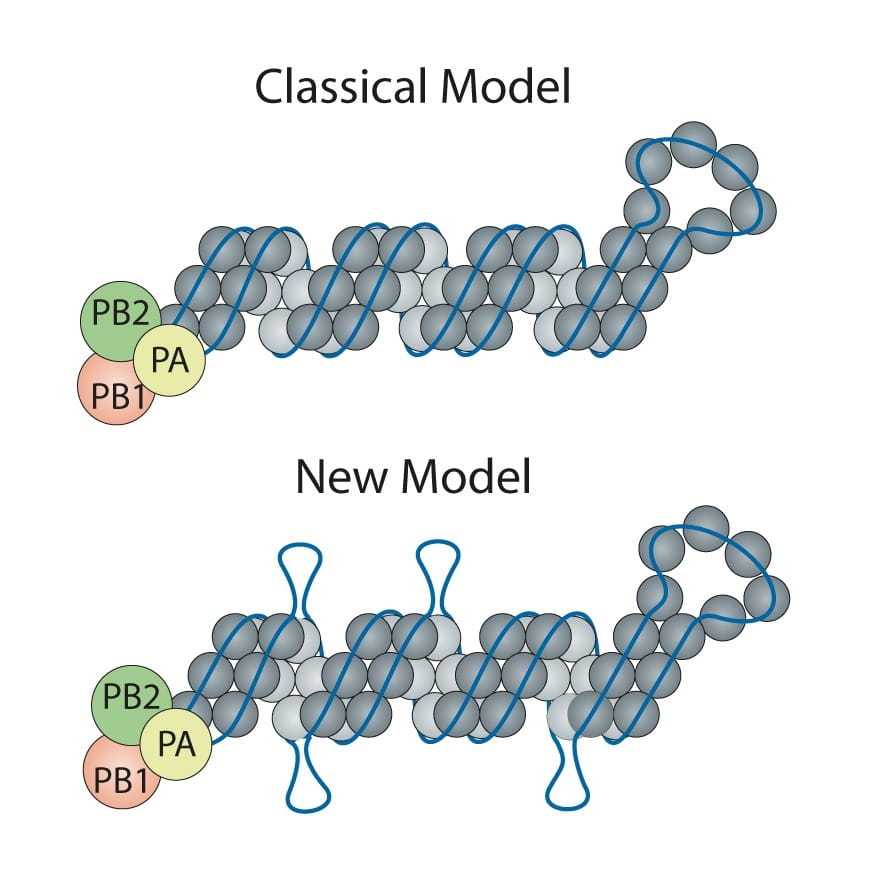
The classic model of the flu virus has these proteins coating the RNA like beads evenly spaced along a string. However, limitations of techniques used in the 1970s when the model was developed meant that unique features—like exposed RNA loops—were lost. Consequently, the universal depiction of influenza in textbooks is of a uniform random binding of proteins along the entire length of each RNA segment.
Lakdawala, who researches how the viruses emerge and spread, teamed up with lead author Nara Lee, Ph.D., assistant professor in Pitt’s Department of Microbiology & Molecular Genetics, who specializes in RNA interactions. The two were curious if there might be any areas along the influenza RNA strand that are more “open” and, therefore, more able to associate with other RNA segments in order to arrive at a package of all eight segments. They used a process called “high-throughput sequencing of RNA by crosslinking immunoprecipitation” (HITS-CLIP) on two strains of influenza A, including the 2009 H1N1 pandemic strain, to get a better understanding for where the proteins bind to the RNA and to see if there were any areas of “naked” RNA.
“Honestly, we didn’t expect to find any since we had all learned the ‘beads on a string’ depiction of viral RNA,” said Lakdawala. “But, amazingly, there are several stretches where the RNA was not bound by the nucleoprotein. This discovery opens up a whole new area of research.”
Contrary to the classic model, Lakdawala and Lee found there are areas of RNA rich with protein coating and others that are exposed and presumably ripe for binding to other viral RNA during reassortment, or the swapping of genomic material between flu viruses. Evolutionary biology expert Vaughn Cooper, Ph.D., associate professor in Pitt’s Department of Microbiology & Molecular Genetics, guided the all-Pitt team to explore how these loops shape virus evolution in nature and during normal flu seasons.
The team is pursuing several potential research opportunities, including predicting the ways different influenza viruses could share genetic material to make new viruses. Knowing this could point scientists to the reassortments most likely to spark a flu pandemic and give public health agencies a leg-up on creating targeted vaccines. There also could be ways to exploit the exposed RNA to make the virus less transmissible and deadly.
“It’s really exciting to suddenly have all these research possibilities open up based on this one discovery,” said Lakdawala. “The reason no one’s uncovered this yet is because we all took for granted that 50-year-old research on the genome architecture, which looked really nice and had an easy explanation, was the full story. It shows that if we don’t constantly resample and question scientific dogma, we could miss a big opportunity.”
Additional authors on this publication are Valerie Le Sage, Ph.D., Adalena V. Nanni, B.S., and Dan J. Snyder, M.S., all of Pitt’s School of Medicine.
This research was funded by the University of Pittsburgh and National Institutes of Health grant U01AI124302.
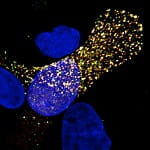
Credit: Seema S. Lakdawala/Pitt
Fluorescent imaging of influenza RNA inside a cell.
Click for high-res image.
Classic vs new flu virus model.
