








7/19/2022
PITTSBURGH - Contrary to early studies, brain levels of enzymes that regulate DNA-folding drop as Alzheimer’s disease progresses, neuroscientists from the University of Pittsburgh, McGill University and Harvard University reported today in Nature Communications.
These dogma-challenging findings, verified across two independent cohorts of live patients with Alzheimer’s disease and on post-mortem brain tissues, show that reduced levels of Histone Deacetylase I (HDAC I)—one of the enzymes that regulate how DNA is packaged inside the cell’s nucleus—are linked to deleterious effects of misfolded beta-amyloid and tau proteins and Alzheimer’s-associated cognitive decline.
Crucially, the new data suggest that using HDAC inhibitors—drugs aimed at reducing HDAC levels that are currently undergoing clinical testing against mild Alzheimer’s disease—might potentially be harming patients rather than helping them. 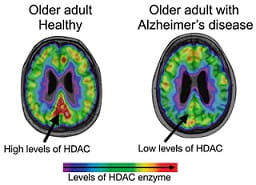
“Approximately one-third of elders who have brain amyloid pathology do not develop Alzheimer’s disease,” said lead author Tharick Pascoal, M.D., Ph.D., assistant professor of psychiatry and neurology in Pitt’s School of Medicine. “Now we have evidence of another factor that dictates whether or not the disease will progress further, and it is related to how the brain environment can affect the expression of our genes.”
Alzheimer’s disease is a fatal neurodegenerative disease that primarily affects the elderly but begins its devious progression decades before symptoms emerge. Forty-four million people live with Alzheimer’s disease or associated dementia. Over time, patients with Alzheimer’s develop cognitive problems—memory loss and difficulty thinking and speaking—that stem from the accumulation of plaques of misfolded beta-amyloid and tangled strands of tau proteins that cause nerve cell death and damage to the brain tissue.
But amyloid and tau pathologies are only one part of the complicated puzzle. Over the last several decades, researchers began to pay attention to other processes—neuroinflammation and changes in the chemical composition of the brain cells’ environment—and how they can affect the course of the disease.
One of these processes is called epigenetic histone modification. By changing the way DNA is folded inside its nucleus—whether it is tightly wound around protein barrels called histones or hung in looser threads—a cell can regulate the efficiency with which genetic information is transcribed into templates for new proteins. This allows the cell to quickly and reversibly tweak the way our genes work and respond to changes in the surrounding environment without altering the DNA sequence itself.
In the long quest to develop safe, effective therapies that stave off cognitive decline and reverse disease progression, a subtype of enzymes that drive epigenetic modifications—HDACs—emerged as promising targets for new Alzheimer’s therapies.
HDACs carry on a chemical reaction that encourages tighter packing of DNA molecules into condensed bundles and restricts biosynthesis of new proteins in response to environmental cues.
Earlier studies on post-mortem brain samples reported that levels of HDACs in the brains of patients with Alzheimer’s increase as the disease progresses. High levels of HDACs were thought to restrict the brain’s ability to produce new functional proteins that make up critical cell components and, therefore, contribute to memory loss and cognitive decline.
The new paper, however, challenges the status quo and adds another piece into an already confusing picture. The loss of HDAC I might be mechanistically linked with the emergence of beta-amyloid and tau pathologies—which, as Pascoal and colleagues showed in their previous paper, are intertwined with brain tissue inflammation and drive Alzheimer’s disease progression—and precede cognitive changes that accompany the disease.
To ensure that their findings represented the real picture across a diverse patient pool, researchers ran two parallel but entirely independent studies at McGill University in Canada and Massachusetts General Hospital, enrolling 94 participants in total. The two sites did not communicate throughout the study period and presented their results unaware of the other group’s findings.
Using a selective molecular tracer called [11C]Martinostat, researchers showed that HDAC I levels were greatly reduced in the brains of people with Alzheimer’s disease compared to non-Alzheimer’s controls, specifically in regions buried deep inside the brain’s core – hippocampus and the midline -- as well as in the brain’s temporal cortex.
Analyses showed that reduced HDAC I in areas of the brain that are most susceptible to Alzheimer’s disease-associated degenerative changes corresponded to higher beta-amyloid and tau burden. It also predicted progressive neurodegeneration and cognitive decline over the two-year period.
While researchers are confident that the rigorous processes that govern clinical trial design ensure the highest standards of patient safety, they warn that efforts to test HDAC inhibitors might be misdirected. Instead, they say, the field needs to further explore the relationship between HDAC activation and disease progression and home in on which specific class of HDACs—out of a total of 18—plays a key role in Alzheimer’s disease-related brain pathology.
Still, the scientists are optimistic.
“The good news is that, by nature, epigenetic processes are changeable,” said Pascoal. “There is a lot of hope for future treatments, and a combination of anti-amyloid therapies with drugs that can rescue the loss of HDACs holds a lot of promise.”
Additional authors of this study are Mira Chamoun, Ph.D., Elad Lax, Ph.D., Hsiao-Ying Wey, Ph.D., Monica Shin, M. Sc, Kok Pin Ng, Ph.D., Min SuKang, Ph.D., SulanthaMathotaarachchi, M.Sc., Andrea Benedet, Ph.D., Joseph Therriault, FirozaLussier, Robert Hopewell, Ph.D., Melissa Savard, Ph.D., Emilie Thomas, Ph.D., Sara Mohaddes, Ph.D., A. Claudio Cuello, Ph.D., Jean-Paul Soucy, Ph.D., GassanMassarweh, Ph.D., HeungsunHwang, Ph.D., Eliane Kobayashi, Ph.D., Marie-Christine Guiot, Ph.D., Moshe Szyf, Ph.D., Serge Gauthier, Ph.D., and Pedro Rosa-Neto, Ph.D., of McGill University; Frederick Schroeder, Ph.D., Jonathan DuBois, Ph.D., BaileighHightower, Ph.D., Tonya Gilbert, Ph.D., Nicole Zürcher, Ph.D., Changning Wang, Ph.D., Bradley Hyman, Ph.D., Bradford Dickerson, Ph.D., and Jacob Hooker, Ph.D., of Massachusetts General Hospital; MallarChakravarty, Ph.D., Sarah Farzin, Ph.D., Alyssa Salaciak, Ph.D., and Stephanie Tullo, Ph.D., of Douglas Mental Health University Institute, Montreal, Canada.
This research was supported by the Alzheimer’s Association (grants AACSF-20-648075, NIRG-12-92090 and NIRP-12-259245), the National Institutes of Health (grants R01AG073267 and R01AG075336), the Weston Brain Institute (grant MOP-11-51-31), Canadian Institutes of Health Research (grant FRN, 152985) and the Fonds de Recherche du Quebec–Sant (FRQS; Chercheur Boursier, no. 2020-VICO-279314).
